
肠道细菌易位的发生机制
2020-07-16肠道是人体最大的细菌及内毒素储存库,肠道微生态环境中约有1*1014个微生物群,包括细菌、病毒、原虫及真菌等,其中最主要为细菌,称为肠道菌群。1966年,Wolochow首先提出细菌易位(bacterial translocation)一词,指原存在于肠腔内的细菌和(或)内毒素,通过某种途径越过肠黏膜屏障,进入肠系膜淋巴结、门静脉系统,继而进入人体循环以及肝、脾、肺等远隔器官的过程。正常情况下,肠道细菌与机体处于动态平衡状态;一旦受到感染、缺血缺氧、严生创伤、手术等刺激,上述平衡状态被打破,可导致多种疾病的发生和发展,甚至引起脓毒症、多器官功能不全乃至多器官功能衰竭。目前人们已广泛认同肠黏膜屏障受损可导致细菌易位这一观点,同时肠道微生态失调和某些重要的蛋白质的作用也逐渐受到了人们的重视。加强对细菌易位机制的研究,对于控制内源性感染、自身感染和内毒素血证,防治肠道相关性疾病,具有重要意义。
一、肠黏膜屏障
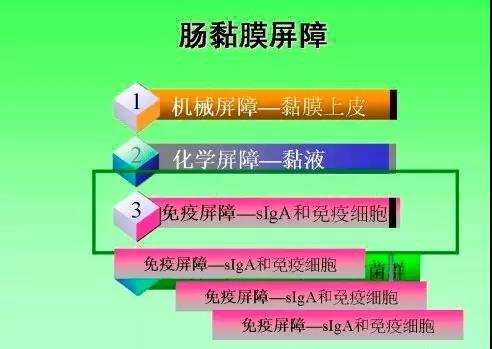
1、肠黏膜的机械屏障和紧密连接:肠黏膜屏障功能(intestinal barrier function)是肠道所具有的特定功能,是指肠上皮具有隔离肠腔内物质,防止致病性抗原入侵的功能,包括机械屏障、微生态屏障、免疫屏障和化学屏障。各种屏障具有不同的结构、不同的分子调控机制和不同的生物学功能,同时又通过各自的信号通路有机结合,共同防御外来抗原物质对机体的侵袭。近年来,研究较为深入的是机械屏障。机械屏障由肠黏膜表面的黏液层、肠上皮本身及其紧密连接等组成,其中紧密连接是基础结构,是由跨膜蛋白、膜周蛋白和调节蛋白组成的多蛋白复合体,跨膜蛋白主要包括密蛋白(claudin)和闭锁蛋白(occludin),膜周蛋白主要是闭锁小带(zonula occluden, ZO)。正常情况下,紧密连接通过调控作用,选择性地转运相应物质,同时有效地阻止肠腔内细菌、毒素等物质的旁细胞转运,维持屏障功能的完整。
2、引起肠道机械屏障功能受损的原因及机制:Deitch曾提出三次打击学说解释肠道机械屏障受损的机制。失血、失液、休克、严重烧伤、呼吸衰竭等应激状态下,机体为保证心、脑等重要器官的血液供应,反射地引起全身血量重新分布,肠道血流量明显减少,产生低灌注,即为第一次打击;肠道缺血再灌注后,中性粒细胞迁移入肠道微循环,淋巴细胞、肠道相关淋巴组织和肠上皮细胞释放炎性因子,形成第二次打击;上述病理变化引起肠上皮细胞因氧化还原功能障碍而坏死,肠道屏障整体性受损,同时紧密连接打开,屏障功能不全,最导致肠细菌和内毒素易位。
此外,目前较公认的引起损伤的原因包括:(1)应激时肠黏膜缺血缺氧,肠上皮细胞有氧代谢障碍,ATP生成减少,细胞膜上的钾-钠泵功能降低,细胞内水钠潴留,引起细胞水肿、坏死,紧密连接结构断裂,肠道屏障功能受损;(2)应激时前炎症因子被激活,并不断循环促进,形成“瀑布”效应,肿瘤坏死因子(TNF)-α、一氧化氮(NO)、血小板活化因子等炎症因子表达增加,紧密连接完整性降低,引起肠黏膜屏障受损;(3)肠黏膜缺血再灌注时,细胞内黄嘌呤氧化酶被大量激活,产生大量氧自由基不能被及时清除,使细胞膜中的多不饱和脂肪酸过氧化,膜的液态性、流动性和通透性发生改变,屏障功能受损。
3、紧密连接打开、肠道机械屏障功能不全,引起细菌易位:紧密连接对调节肠屏障的通透性、防止细菌内毒素及毒性大分子物质进入体内具有重要意义。目前发现至少可通过两种途径维持紧密连接的通透性。渗漏途径(the leak pathway)允许细菌脂多糖等大分子的细胞旁转运,但受分子大小选择性制约,不能通过完整的细菌整体。另一途径则由密蛋白形成小孔结构,受病原体或其他刺激时小也结构发生改变,使肠道通透性增加。
细菌侵袭肠黏膜上皮细胞时,TNF等细胞因子表达增加,引起肌球蛋白轻链激酶(myosin light chain kinase, MLCK)表达增加,肌球蛋白轻链磷酸化后,肌动蛋白ATP酶活化,肌动蛋白肌球蛋白环收缩;同时白细胞介素(IL)-13表达增加,上皮细胞凋亡,密蛋白表达,上皮完整性受损,紧密连接打开,肠道通透性增加,肠道屏障功能不全。肠致病型大肠杆菌可分泌EspF、EspG和Map,调节小Rho样G蛋白使肌动蛋白肌丝聚合,紧密连接打开,降低对细菌毒素跨膜转运的抵抗力,并在宿主细胞表面提供细菌定植的基座,破坏肠道屏障,肠道通透性增加,因此有利于细菌易位。但需要注意的是,人们发现在克罗恩病患者健康家属的肠黏膜中,紧密连接蛋白表现出了同样的改变,表明仅仅紧密连接通透性增加并不足以致病,MLCK活化后还需要引起黏膜免疫细胞的激活。敲除MLCK表达基因的小鼠结肠炎较野生小鼠发展更快、更严重,而其他肠上皮缺陷引起的反应却并不明显,提示MLCK活化在紧密连接信号通路中的重要作用,把MLCK活化后引起的紧密连接通透性增加作为靶点能够加速免疫介导的炎症反应的启动。Smith等还提出紧密连接蛋白可能通过与其他蛋白分子协同作用参与细菌易位。HIV病毒感染时,claudin-2蛋白表达增加,正常的紧密连接结构被打乱,屏障结构被破坏,使细菌得以与细胞侧面的黏附分子整合素跨膜粘结蛋白聚糖家族成员syndecan-1接触并结合,促进细菌易位,并引起全身性感染,提示claudin-2可通过syndecan-1作用来易化细菌易位。
二、肠道微生态失调引起肠道细菌易位
肠道的正常菌群由高密度的原籍菌群和部分低密度的外籍菌群及环境菌群构成,并按一定数量和比例分布在肠道的不同节段和部位,参与宿主的代谢、免疫、生化和生物拮抗等多方面的作用,以维持微生态平衡。肠道细菌过度繁殖时,肠道微生态失调,使黏附到肠壁的细菌增多,同时定植机会增加,产生大量代谢产物和毒素,破坏肠黏膜结构,使肠道通透性增加,引起细菌易位。Guarner和Soriano总结近年文献后认为,肠道通过时间延长、细菌过度生长是肝硬化患者发生细菌易位的机制之一。肝硬化患者胆汁酸缺乏,免疫力低下,肠道细菌过度繁殖;同时患者肠道通透性增加,肠道内胆汁酸被动吸收增加;过多繁殖的细菌又能加强胆汁酸的去结合作用,加剧了肠道内胆汁酸的缺乏,形成了进一步加剧肠道内细菌繁殖的恶性循环,最终引起细菌易位。此外,核受体法尼醇受体(farnesoid X receptor)与肠黏膜上皮细胞的跨膜蛋白syndecan-1结合,也可影响胆汁酸代谢,抑制肠道细菌过度生长,并且避免了肠道黏膜表面的断裂、肠道上皮的水肿以及肠上皮绒毛的中性粒细胞浸润,保护了肠黏膜屏障的完整性,从而减少细菌易位。细菌易位后引起内源性NO和炎性因子TNF-α、IL-12分泌增加,NO合成增加和宿主免疫防御机制紊乱均能促使肠通透性增加和黏膜损伤,又能够促进细菌易位,形成恶性循环,同时还可引起全身血管阻力降低,平均动脉压降低,并且肝静脉压力梯度增加、血管舒张功能受损,严重影响预后。使用选择性肠道清洁剂(SDD)可选择性地根除口咽部及肠道的潜在致病菌,同时保留肠道固有的专性厌氧菌,从而维持机体的定植抗力,抑制细菌易位和潜在致病菌的感染。一项前瞻性的随机对照临床试验证明,SDD能够有效降低重症监护室患者耐万古霉素肠球菌和耐甲氧西林金黄色葡萄球菌的定植,从而抑制细菌易位,并显著降低病死率。使用益生菌和益生元制剂能直接补充机体的正常菌群或选择性刺激正常菌群的生长繁殖,从而竞争性抑制外细菌的定植和内源性条件致病菌的过度生长,有效抑制肠道微生态失调,减少细菌易位的发生。
三、参与肠道细菌易位的细胞表面蛋白和细胞外基质
细菌与宿主细胞不同的细胞表面蛋白或细胞外基质通过受体-配体作用结合在细菌易位过程中具有重要意义,其中参与细菌易位的细胞表面蛋白主要是跨膜蛋白等,细胞外基质成分主要是层粘连蛋白等。目前报道较多的主要有以下几种:
1、Toll样受体(Toll-like receptors, TLR):TLR是表达于肠上皮细胞表面的模式识别受体(pattern recognition receptors, PRR)。TLR胞外段参与对细菌脂蛋白、脂磷壁酸、鞭毛等结构的识别;胞内段含有与IL-1R的胞质区结构相似的TIR结构域,是TLR向下游进行信号传导的基本元件。TLR与相应配体结合后活化,髓系分化因子88(myeloid differentiation factor 88, My88)等效应分五与TIR结合,募集下游信号分子IRAK-4,IRAK-4磷酸化后激活IRAK-1,IRAK-1离开受体配体复合物、与下游的接头分子TRAF6可泛素化降解IKK复合体,激活TGF-β激酶继而催化IKK-B磷酸化,最终激活核因子(NF)-κB信号通路,引起炎性因子表达。Kang等还发现,细菌或其组分与TLRS结合后诱导MAPK家族的p38α表达,引起趋化因子表达,T细胞募集。上述改变均可影响机体正常的免疫防御机制,使机体不能有效地遏制和消除易位的细菌和内毒素,是促发细菌易位的最重要因素。
2、NOD样受体(NLR):NOD样受体是一种胞质识别受体,它和TLR在不同部位特异性识别共生菌和致病菌并相互影响。NOD1和NOD2是NLR家族中最典型的成员,主要与细菌肽聚糖结合,激活后引起下游RIP2分子募集,激活(NF)-κB、MAPK与caspase-1等不同途径,诱导前炎症因子和趋化因子表达以及DC的活化,参与调节机体的免疫应答和细菌易位。
3、Syndecan-1分子:Syndecan-1是表达于肠黏膜上皮细胞的跨膜糖蛋白,属于粘附分子整合素跨膜粘结蛋白(heparan sulfate proteoglycan, HSPG)家族成员,作为共受体调节细胞与胃肠道微环境之间的反应,在细菌感染和炎症的发生与发展中有着重要作用。正常情况Syndecan-1能够限制肠道共生菌对肠道的粘附,维持肠道菌群平衡;受到各种病理因素刺激时,Syndecan-1发挥受体作用,结合细菌表达的乙酰肝素结合分子,介导致病菌粘附宿主细胞,诱导宿主细胞肌动蛋白聚合,使致病菌被内化后侵入宿主细胞并引起细菌易位。奈瑟菌表达的外膜蛋白OpaHSPG结合高表达Syndecan-1的海拉上皮细胞后,可使黏附能力和侵袭能力同时提高3倍;敲除Syndecan-1胞内段表达基因,细菌侵袭能力显著降低,但粘附能力未见明显改变;而用乙酰肝素酶酶解HS链或使用HS类似物乙酰肝素直接处理宿主细胞,均可抑制细菌的粘附。
4、粘蛋白(mucin)和肠三叶肽(trefoil3 factor, TFF3):粘蛋白由杯状细胞分泌, 是黏液最主要的组成蛋白,能够在肠上皮表面形成一层深达150um的连续性凝胶样的多糖网络阻挡细菌等大分子抗原的侵入。粘蛋白层分为内外2层。外层粘蛋白暴露的化学基团与肠上皮表面结构类似,易于与细菌表面鞭毛等多糖-蛋白质附件相互作用,为细菌提供粘附部位。粘蛋白可通过与致病菌竞争粘附受体,抑制致病菌在肠道的粘附定植。粘附之后,粘蛋白协同分泌型IgA形成抗感染的抗体屏障,通过肠道蠕动将细菌清除。内层粘蛋白可在上皮表面形成保护区,维持上皮细胞分泌的抗菌肽的完整性,并抑制抗菌肽播散入肠腔,以维持内层黏液处于近乎无菌的水平。Bergstrom等通过实验发现,粘蛋白MUC2能够清除松散粘附的细菌群负担、限制细菌移位,发挥保护作用。TFF3也是由杯状细胞特异分泌的小分子多肽,能同粘蛋白的糖链结合,形成稳定的凝胶复合物,维护胃肠黏液层,介导肠上皮迁移、促进受损肠黏膜修复。大肠杆菌感染时,TLR2配体Pam3CSK4可诱导TFF3表达,减少结直肠炎中的直肠出血、改善组织学情况、减少细胞凋亡并加强紧密连接蛋白ZO1的连接,并抑制细菌易位。
5、其他可能有关的蛋白分子:近年来,人们还发现,以往主要见于中性粒细胞表面的甲酰基肽受体家族成员(formylated peptide receptors, FPR)也可表达于肠上皮细胞,参与对细菌及其组分的识别并抑制细菌易位。某些类杆菌等非致病菌可通过与过氧化物酶体增殖因子活化受体γ(peroxisome proliferators-activated receptor gamma, PPARγ)相互作用,降低炎性介质的表达,避免宿主免疫防御机制紊乱,而抑制细菌易位。革兰阳性菌的脂磷壁酸能结合细胞表面G蛋白的共受体血小板活化受体(platelet-activating factor, PAF receptor),激活Ras/Raf/MEK/NF-κB等信号通路和黏蛋白MUC2转录,参与细菌易位。
自1996年首创细菌易位一词到现在,胃肠微生态引起了越来越多的关注。近半个世纪以来,肠道细菌易位相关的疾病谱在不断扩展,炎症性肠病、肠易激综合征、肝硬化、多器官功能障碍综合征、自发性腹膜炎、慢性肾功能衰竭、冠状动脉性心脏病、多发性硬化等都与肠道细菌易位有关。目前可导致细菌易位的相关机制逐渐得到大家的重视,但具体的分子机制和信号通路尚未阐明,还需要进行更深入的研究,以期为防治细菌易位相关疾病提供新思路和新方法。
温馨提示:因患者情况存在差异,以上内容仅供参考,具体诊疗请咨询医生指导。
-
肠道细菌易位的发生机制
肠道是人体最大的细菌及内毒素储存库,肠道微生态环境中约有1*1014个微生物群,包括细菌、病毒、原虫及真菌等,其中最主要为细菌,称为肠道菌群。1966...
-
肠道菌群失调的原因
肠道菌群失调的原因?健康人的胃肠道内寄居着种类繁多的微生物,这些微生物称为肠道菌群。肠道菌群按一定的比例组合,各菌间互相制约,互相依存,在...
-
肠道菌群失调诊断治疗建议
正常情况下,肠道菌群在体内与外部环境保持着动态平衡,并对人体的健康起着重要作用。如果这种平衡在某些情况下被打破,便形成肠道菌群失调(intestina...
-
要健康,先伺候好你的肠道菌群这很重要
越来越多的研究证明,肠道菌群对人体健康和生活起着复杂而重要的作用。例如阻止病原菌的入侵和繁殖;通过若干单分子和代谢产物调节免疫系统;帮助小肠微...
-
如何确定肠道菌群失调及其检测方法
如何确定肠道菌群失调及其检测方法?肠道菌群失调系指肠道正常细菌间的比例发生重大变化并引起某些临床症状而言。正常情况时肠道各种非致病菌间有...
-
数据告诉你,肠道菌群平衡有多重要!
我们都知道,地球生态平衡对于整个地球环境是至关重要的。同样,肠道微生态平衡对于我们的机体健康也至关重要。在人肠道内至少共生着1000种、数量高达...